
发布时间:2026-06-10

标题:PRDM9 Deficiency Drives Mosaic Promoter Deletions in Sporadic Hirschsprung Disease and Supports Blood-based Molecular Stratification
期刊:Gastroenterology(IF28.7,中国科学院1区TOP)
DOI:10.1053/j.gastro.2026.05.020
发表时间:2026年6月5日
研究背景
先天性巨结肠(HSCR)是一种严重的先天性肠神经发育障碍性疾病,多见于新生儿或婴幼儿,以肠道远端神经节细胞缺失为特征,主要表现为肠道功能障碍,可导致严重肠梗阻。尽管已有研究发现RET、EDNRB等多个致病基因,但这些突变仅能解释少数家族性病例,而80-90%的散发性HSCR病例仍无法解释。因此,亟需探究非经典致病机制,并开发精准的诊断工具。
关键发现
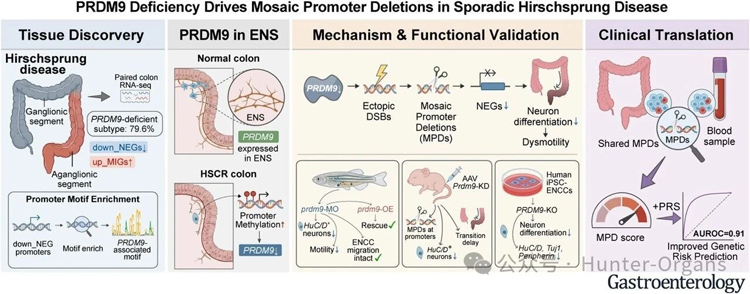
本研究利用斑马鱼、小鼠和人诱导多能干细胞(iPSCs)来源的肠神经嵴细胞模型等,揭示了PRDM9缺陷的致病机制及转化价值,发现PRDM9缺失可驱动神经发生相关基因启动子区域发生嵌合性缺失,损害肠神经元分化,并支持基于外周血的分子分层。
主要分子亚群的鉴定:通过对103例散发性先天性巨结肠的研究发现,约80%的散发性HSCR患者存在神经发生相关基因的协同抑制,表明存在共同的致病通路;
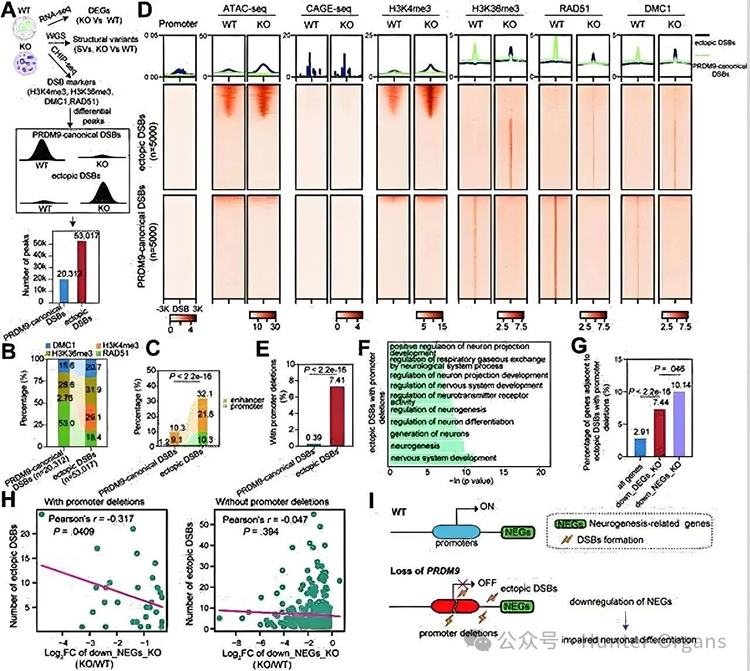
PRDM9缺陷导致疾病发生:PRDM9基因在正常肠神经系统中表达,但在患者无神经节肠段中因启动子超甲基化而显著下调。PRDM9缺失导致DNA双链断裂异常聚集在启动子和增强子区,并产生嵌合型启动子缺失(MPDs),直接导致肠神经元分化;
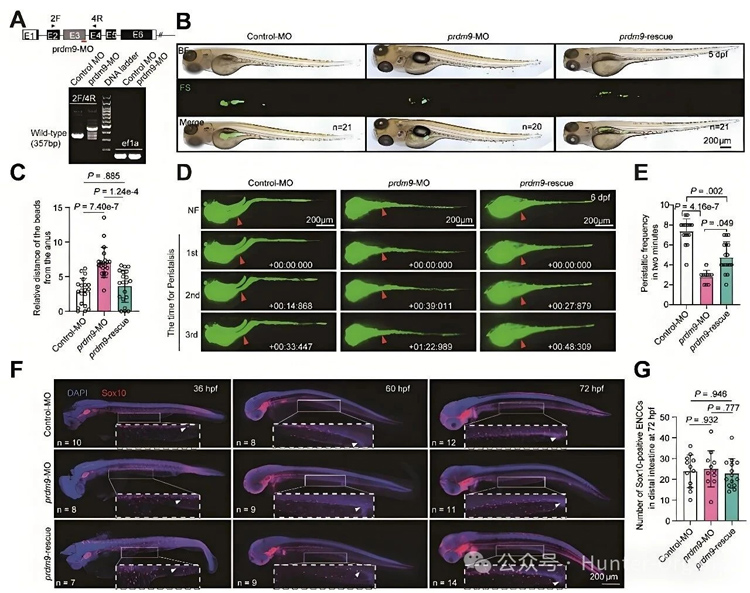
多模型功能验证:在斑马鱼、小鼠和人诱导多能干细胞(iPSCs)来源的肠神经嵴细胞模型中,PRDM9功能缺失都会引起HuC/D⁺肠神经元数量减少、分化障碍及肠道运动功能受损;
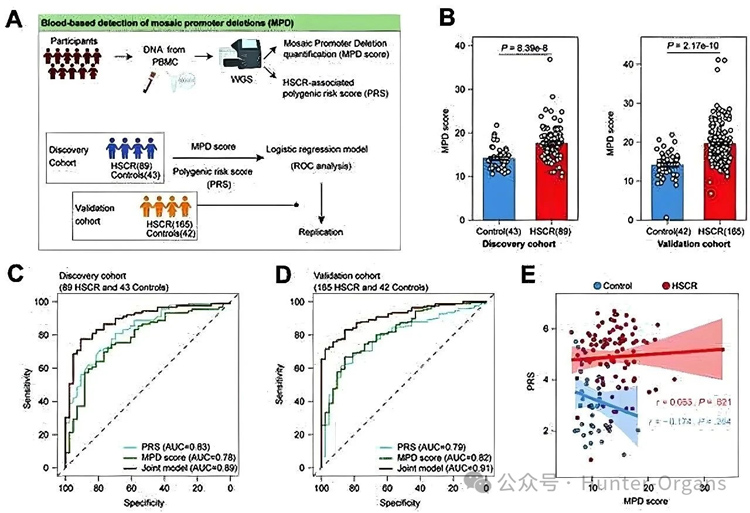
无创分子分层新工具:基于外周血中可检测的MPD构建评分模型,在发现队列(AUC 0.78)和独立验证队列(AUC 0.82)中均能有效区分HSCR患者与对照;联合多基因风险评分后AUC分别提升至0.89和0.91,实现了散发性HSCR的非侵入性分子诊断与分层。
临床意义
填补病因空白:揭示了PRDM9缺陷是散发性HSCR中占比约80%的主要分子亚群的致病机制,为大多数既往无法解释的病例提供了分子病因;
无创诊断新工具:基于血液的MPD评分可作为非侵入性生物标志物,联合PRS后可显著提升诊断性能,有望用于HSCR的早期筛查和精准分层;
机制启发:PRDM9缺陷通过诱导启动子区域异常DNA断裂及嵌合缺失损害肠神经元分化,拓展了对神经嵴疾病致病机制的认识。
上一条:益生菌功效从概念到实证丨环特多维循证功效技术平台亮相益生菌与健康研讨会
下一条:没有了!






